Consequences of absent liver enzyme in children with PKU would result to deficient tyrosine leading to the following conditions:
- Absence of serotonin, dopamine and epinephrine
Result: Faulty nerve (Nervous System) transmission
Neurotransmitters communicate impulses to the nerve cells. Lack of tyrosine would lead to deterioration of this function. Mood regulation is also connected to the presence of these chemicals (dopamine, serotonin, and epinephrine); therefore, alteration of one’s disposition and temperament will be expected.
- Deficient Melanin levels
Result: Unusual skin color
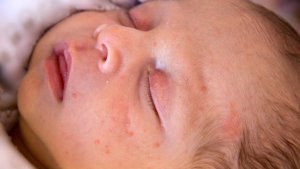
Melanin is responsible for skin pigmentation. Deficient levels of melanin lead to a very fair complexion, a light blond hair and blue eyes.
- Hyposecretion of thyroid hormones
Result: Permanent brain damage (Mental Retardation) and developmental delay
Thyroid glands are located at the throat that comprises the two lateral masses on each side of the trachea. Before the two active thyroid hormones are produced, a process known as iodide trapping (iodide ion is concentrated within the thyroid) occurs. Then iodide is dissolved inside the follicular cells of the thyroid to become iodine and later released as a colloid. Colloids contain thyroglobulins which are made up of the amino acid tyrosine. Iodide when combined with tyrosine produces Monoiodotyrosine (MIT) and Diiodotyrosine (DIT). Conversion of MIT and DIT would form the two active thyroid hormone, triiodothyronine (T3) and Thyroxine (T4). These hormones are stored in the follicular cells until needed. T3 and T4 are primarily responsible for cellular metabolism affects nearly all cells in the body. They play a vital role for normal development to occur.
In PKU, no Monoiodotyrosine (MIT) and Diiodotyrosine (DIT) is formed due to absence of tyrosine. Production of T3 and T4 would be inevitable causing decrease basal metabolism, cessation of cognitive and physical development. Most children with PKU are cognitively challenged having an IQ of less than 20.
- Increase Phenylalanine levels
Result: Mousy urine odor
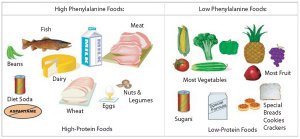
Phenylalanine levels increase due to the absence of the liver enzyme. The end product of phenylalanine metabolism is phenylpyruvic acid (a keto acid). The by-product spills into the urine that gives it a strong “mousy” or “musty” odor that often spreads through the entire body of the infant or child. This is the reason why the disorder is called phenylketonuria (meaning there is phenylpruvic or keto acid in the urine)